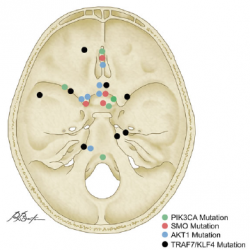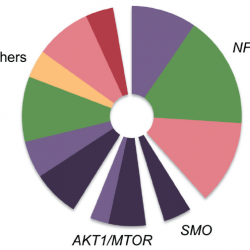
Papillary craniopharyngioma (PCP) is an intracranial tumor that results in high levels of morbidity.We recently demonstrated that the vast majority of these tumors harbor the oncogenic BRAF V600E mutation. The pathologic diagnosis of PCP can now be confirmed using mutation specific immunohistochemistry and targeted genetic testing. Treatment with targeted agents is now also a possibility in select situations. We recently reported a patient with a multiply recurrent PCP in whom targeting both BRAF and MEK resulted in a dramatic therapeutic response with a marked anti-tumor immune response. This work shows that activation of the MAPK pathway is the likely principal oncogenic driver of these tumors.We will now investigate the efficacy of this approach in a multicenter phase II clinical trial. Post-treatment resection samples will be monitored for the emergence of resistance mechanisms. Further advances in the non-invasive diagnosis of PCP by radiologic criteria and by cell-free DNA testing could someday allow neo-adjuvant therapy for this disease in select patient populations.