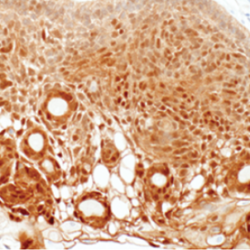
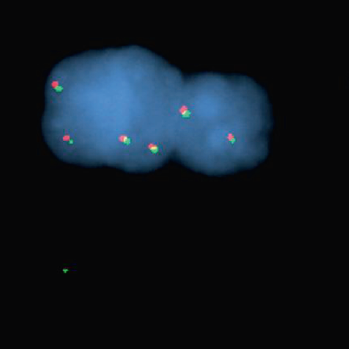
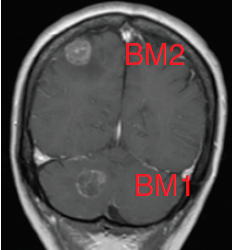
Glioblastomas are malignant neoplasms composed of diverse cell populations. This intratumoral diversity has an underlying architecture, with a hierarchical relationship through clonal evolution from a common ancestor. Therapies are limited by emergence of resistant subclones from this phylogenetic reservoir. To characterize this clonal ancestral origin of recurrent tumors, we determined phylogenetic relationships using whole exome sequencing of pre-treatment IDH1/2 wild-type glioblastoma specimens, matched to post-treatment autopsy samples (n = 9) and metastatic extracranial post-treatment autopsy samples (n = 3). We identified “truncal” genetic events common to the evolutionary ancestry of the initial specimen and later recurrences, thereby inferring the identity of the precursor cell population. Mutations were identified in a subset of cases in known glioblastoma genes such as NF1(n = 3), TP53(n = 4) and EGFR(n = 5). However, by phylogenetic analysis, there were no protein-coding mutations as recurrent truncal events across the majority of cases. In contrast, whole copy-loss of chromosome 10 (12 of 12 cases), copy-loss of chromosome 9p21 (11 of 12 cases) and copy-gain in chromosome 7 (10 of 12 cases) were identified as shared events in the majority of cases. Strikingly, mutations in the TERT promoter were also identified as shared events in all evaluated pairs (9 of 9). Thus, we define four truncal non-coding genomic alterations that represent early genomic events in gliomagenesis, that identify the persistent cellular reservoir from which glioblastoma recurrences emerge. Therapies to target these key early genomic events are needed. These findings offer an evolutionary explanation for why precision therapies that target protein-coding mutations lack efficacy in GBM.
Brastianos PK, Nayyar N, Rosebrock D, Leshchiner I, Gill CM, Livitz D, Bertalan MS, D’Andrea M, Hoang K, Aquilanti E, Chukwueke UN, Kaneb A, Chi A, Plotkin S, Gerstner ER, Frosch MP, Suva ML, Cahill DP, Getz G, Batchelor TT.
NPJ Precis Oncol. 2017 Sep 18;1(1):33. doi: 10.1038/s41698-017-0035-9. eCollection 2017.